"DNADynamo is by far and away the best and quickest software for checking sequences"- Professor Greg Towers, University College London
A guide to basic DNA Dynamo sequence analysis functions.
So you've sent off some DNA to be sequenced and received back some .abi or .scf files containing the trace data and base calls. Now you want to analyse your sequencing data. DNA Dynamo can help you accomplish this quickly and efficiently, without the need to spend hours analysing each separate data trace manually. By auto assembling translated multiple alignments of your sequencing data, where each base in the alignment is linked to it's chromatogram peak, and presenting an interactive graphic map of sequencing discrepancies, open reading frames and features maps, as well as interfacing with NCBIs Blast and VecScreen services, you can get a handle on your sequence data within minutes. Intuitive sequence editing buttons allow you to manually edit your chromatogram trace base calls and then save the edited and assembled sequence for future reference. By spending a few minutes working out how to use DNA Dynamo for sequence analysis you will save yourself a vast amount of time and effort in the future. DNA Dynamo works in two modes for sequence assembly, depending on whether you know what the expected sequence should be (eg sequencing a construct after subcloning or confirming the result of a site directed mutagenesis experiment) or the sequence is unknown (eg sequencing inserts from a two-hybrid screen, degenerate PCR or other similar library screens).




This video will introduce you to some of the basic sequencing functions in DNADynamo. You can also read a text based introduction below,
which covers some additional options.
A) Guide/Reference Mode - to confirm your sequence against the expected result or find
mutations.
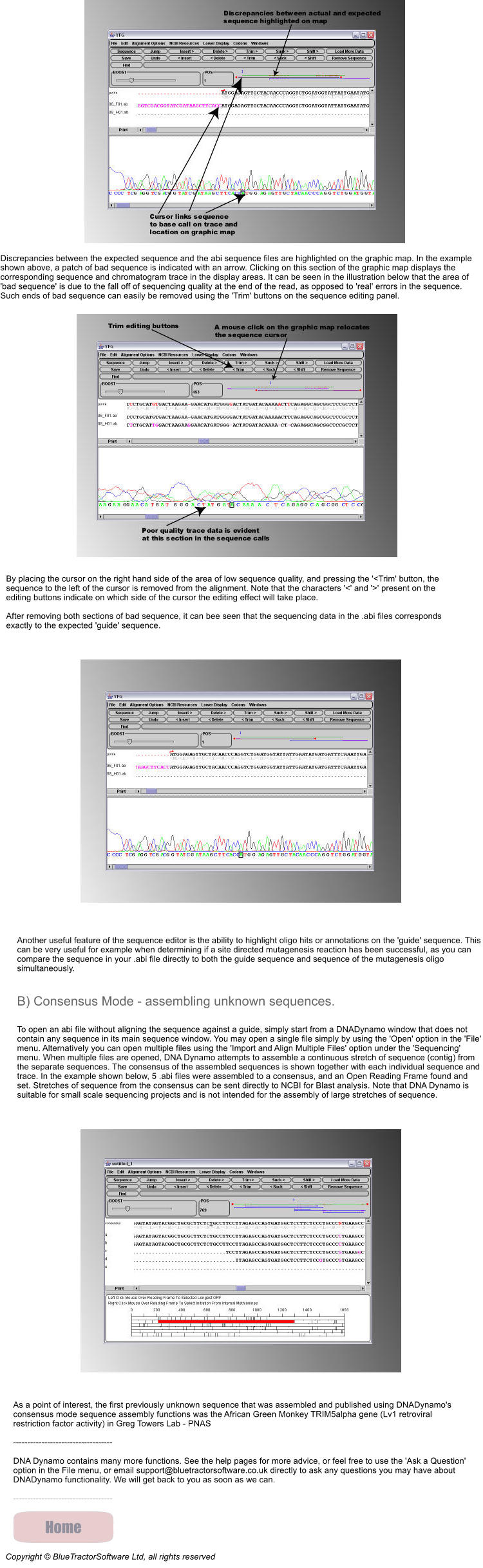
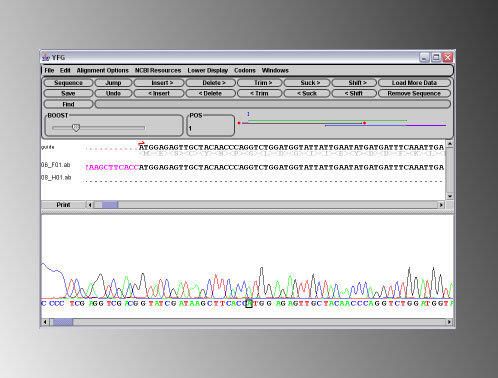
For example, you have just PCR sub-cloned Your Favorite Gene (YFG) into a vector, and you need to confirm that no errors have been introduced during the PCR reaction, so you have sent of a few minipreps to your sequencing service together with sequencing primers to sequence in from both ends, and received back a bunch of .abi files. 1) open a DNA Dynamo file that contains the wildtype sequence of YFG in the main sequence window. From the 'Sequencing' menu, select 'Open And Align Sequence Data Files to this Windows Sequence', and then select the .abi files that represent the sequence of one clone. DNA Dynamo will align the sequences in the .abi files to the sequence of the 'guide' dna in the main sequence window, and display the aligned sequences in a sequencing editor window. The result of such an alignment, where two .abi files were aligned against expected sequence, is shown below. Note that DNA Dynamo automatically determines whether the sequence in the abi files is better aligned as forward or reverse - and this is indicated on the graphics map by arrows - though these are not visible at this resolution.
•
Windows
•
OSX
•
Linux

DNADynamo
BlueTractorSoftware















"DNADynamo is by far and away the best and quickest software for checking sequences"- Professor Greg Towers, University
College London
A guide to basic DNA Dynamo sequence analysis functions.
So you've sent off some DNA to be sequenced and received back some .abi or .scf files containing the trace data and base calls. Now you want to analyse your sequencing data. DNA Dynamo can help you accomplish this quickly and efficiently, without the need to spend hours analysing each separate data trace manually. By auto assembling translated multiple alignments of your sequencing data, where each base in the alignment is linked to it's chromatogram peak, and presenting an interactive graphic map of sequencing discrepancies, open reading frames and features maps, as well as interfacing with NCBIs Blast and VecScreen services, you can get a handle on your sequence data within minutes. Intuitive sequence editing buttons allow you to manually edit your chromatogram trace base calls and then save the edited and assembled sequence for future reference. By spending a few minutes working out how to use DNA Dynamo for sequence analysis you will save yourself a vast amount of time and effort in the future. DNA Dynamo works in two modes for sequence assembly, depending on whether you know what the expected sequence should be (eg sequencing a construct after subcloning or confirming the result of a site directed mutagenesis experiment) or the sequence is unknown (eg sequencing inserts from a two-hybrid screen, degenerate PCR or other similar library screens).




This video will introduce you to some of the basic sequencing functions in DNADynamo. You can also read a text based introduction
below, which covers some additional options.
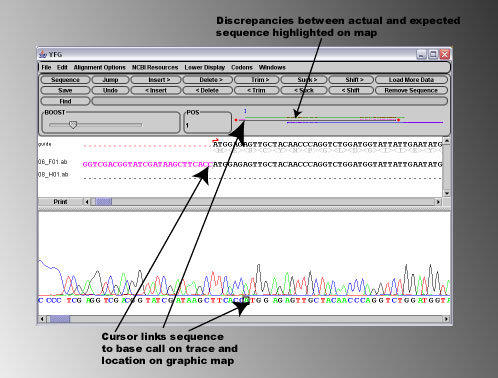
Discrepancies between the expected sequence and the abi sequence files are highlighted on the graphic map. In the example
shown above, a patch of bad sequence is indicated with an arrow. Clicking on this section of the graphic map displays the
corresponding sequence and chromatogram trace in the display areas. It can be seen in the illustration below that the area of
'bad sequence' is due to the fall off of sequencing quality at the end of the read, as opposed to 'real' errors in the sequence.
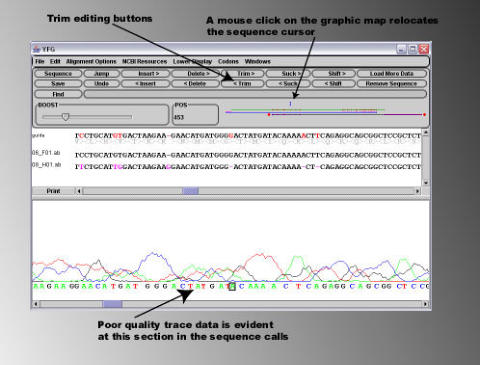
Such ends of bad sequence can easily be removed using the 'Trim' buttons on the sequence editing panel.
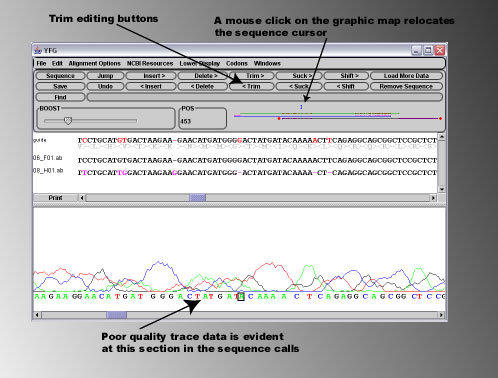
By placing the cursor on the right hand side of the area of low sequence quality, and pressing the '<Trim' button, the
sequence to the left of the cursor is removed from the alignment. Note that the characters '<' and '>' present on the
editing buttons indicate on which side of the cursor the editing effect will take place.
After removing both sections of bad sequence, it can bee seen that the sequencing data in the .abi files corresponds
exactly to the expected 'guide' sequence.
Another useful feature of the sequence editor is the ability to highlight oligo hits or annotations on the 'guide' sequence. This
can be very useful for example when determining if a site directed mutagenesis reaction has been successful, as you can
compare the sequence in your .abi file directly to both the guide sequence and sequence of the mutagenesis oligo
simultaneously.
B) Consensus Mode - assembling unknown sequences.
To open an abi file without aligning the sequence against a guide, simply start from a DNADynamo window that does not contain any sequence in its main sequence window. You may open a single file simply by using the 'Open' option in the 'File' menu. Alternatively you can open multiple files using the 'Import and Align Multiple Files' option under the 'Sequencing' menu. When multiple files are opened, DNA Dynamo attempts to assemble a continuous stretch of sequence (contig) from the separate sequences. The consensus of the assembled sequences is shown together with each individual sequence and trace. In the example shown below, 5 .abi files were assembled to a consensus, and an Open Reading Frame found and set. Stretches of sequence from the consensus can be sent directly to NCBI for Blast analysis. Note that DNA Dynamo is suitable for small scale sequencing projects and is not intended for the assembly of large stretches of sequence.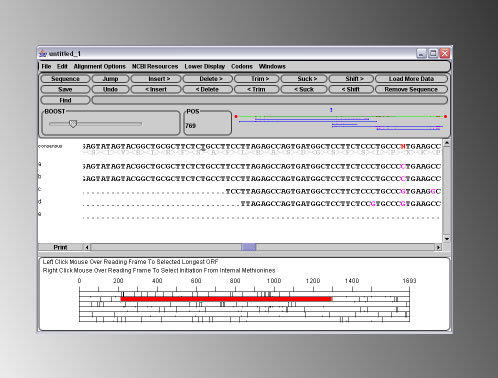
As a point of interest, the first previously unknown sequence that was assembled and published using DNADynamo's
consensus mode sequence assembly functions was the African Green Monkey TRIM5alpha gene (Lv1 retroviral
restriction factor activity) in Greg Towers Lab - PNAS
-----------------------------------
DNA Dynamo contains many more functions. See the help pages for more advice, or feel free to use the 'Ask a Question'
option in the File menu, or email support@bluetractorsoftware.co.uk directly to ask any questions you may have about
DNADynamo functionality. We will get back to you as soon as we can.
-----------------------------------
Copyright © BlueTractorSoftware Ltd, all rights reserved
A) Guide/Reference Mode - to confirm your sequence against the expected result or
find mutations.
For example, you have just PCR sub-cloned Your Favorite Gene (YFG) into a vector, and you need to confirm that no errors have been introduced during the PCR reaction, so you have sent of a few minipreps to your sequencing service together with sequencing primers to sequence in from both ends, and received back a bunch of .abi files. 1) open a DNA Dynamo file that contains the wildtype sequence of YFG in the main sequence window. From the 'Sequencing' menu, select 'Open And Align Sequence Data Files to this Windows Sequence', and then select the .abi files that represent the sequence of one clone. DNA Dynamo will align the sequences in the .abi files to the sequence of the 'guide' dna in the main sequence window, and display the aligned sequences in a sequencing editor window. The result of such an alignment, where two .abi files were aligned against expected sequence, is shown below. Note that DNA Dynamo automatically determines whether the sequence in the abi files is better aligned as forward or reverse - and this is indicated on the graphics map by arrows - though these are not visible at this resolution.







"DNADynamo is by far and away the best and quickest software for checking
sequences"- Professor Greg Towers, University College London
A guide to basic DNA Dynamo
sequence analysis functions.
So you've sent off some DNA to be sequenced and received back some .abi or .scf files containing the trace data and base calls. Now you want to analyse your sequencing data. DNA Dynamo can help you accomplish this quickly and efficiently, without the need to spend hours analysing each separate data trace manually. By auto assembling translated multiple alignments of your sequencing data, where each base in the alignment is linked to it's chromatogram peak, and presenting an interactive graphic map of sequencing discrepancies, open reading frames and features maps, as well as interfacing with NCBIs Blast and VecScreen services, you can get a handle on your sequence data within minutes. Intuitive sequence editing buttons allow you to manually edit your chromatogram trace base calls and then save the edited and assembled sequence for future reference. By spending a few minutes working out how to use DNA Dynamo for sequence analysis you will save yourself a vast amount of time and effort in the future. DNA Dynamo works in two modes for sequence assembly, depending on whether you know what the expected sequence should be (eg sequencing a construct after subcloning or confirming the result of a site directed mutagenesis experiment) or the sequence is unknown (eg sequencing inserts from a two-hybrid screen, degenerate PCR or other similar library screens).



This video will introduce you to some of the basic sequencing functions in
DNADynamo. You can also read a text based introduction below, which covers
some additional options.

Discrepancies between the expected sequence and the abi sequence files are
highlighted on the graphic map. In the example shown above, a patch of bad
sequence is indicated with an arrow. Clicking on this section of the graphic map
displays the corresponding sequence and chromatogram trace in the display
areas. It can be seen in the illustration below that the area of 'bad sequence' is
due to the fall off of sequencing quality at the end of the read, as opposed to 'real'
errors in the sequence. Such ends of bad sequence can easily be removed using
the 'Trim' buttons on the sequence editing panel.
By placing the cursor on the right hand side of the area of low sequence quality,
and pressing the '<Trim' button, the sequence to the left of the cursor is removed
from the alignment. Note that the characters '<' and '>' present on the editing
buttons indicate on which side of the cursor the editing effect will take place.
After removing both sections of bad sequence, it can bee seen that the
sequencing data in the .abi files corresponds exactly to the expected 'guide'
sequence.

Another useful feature of the sequence editor is the ability to highlight oligo hits or
annotations on the 'guide' sequence. This can be very useful for example when
determining if a site directed mutagenesis reaction has been successful, as you
can compare the sequence in your .abi file directly to both the guide sequence
and sequence of the mutagenesis oligo simultaneously.
B) Consensus Mode - assembling unknown sequences.
To open an abi file without aligning the sequence against a guide, simply start from a DNADynamo window that does not contain any sequence in its main sequence window. You may open a single file simply by using the 'Open' option in the 'File' menu. Alternatively you can open multiple files using the 'Import and Align Multiple Files' option under the 'Sequencing' menu. When multiple files are opened, DNA Dynamo attempts to assemble a continuous stretch of sequence (contig) from the separate sequences. The consensus of the assembled sequences is shown together with each individual sequence and trace. In the example shown below, 5 .abi files were assembled to a consensus, and an Open Reading Frame found and set. Stretches of sequence from the consensus can be sent directly to NCBI for Blast analysis. Note that DNA Dynamo is suitable for small scale sequencing projects and is not intended for the assembly of large stretches of sequence.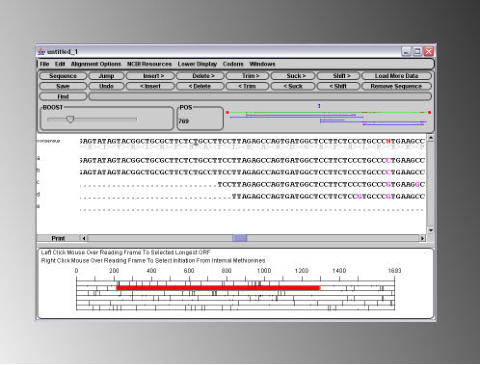
As a point of interest, the first previously unknown sequence that was assembled
and published using DNADynamo's consensus mode sequence assembly
functions was the African Green Monkey TRIM5alpha gene (Lv1 retroviral
restriction factor activity) in Greg Towers Lab - PNAS
-----------------------------------
DNA Dynamo contains many more functions. See the help pages for more advice,
or feel free to use the 'Ask a Question' option in the File menu, or email
support@bluetractorsoftware.co.uk directly to ask any questions you may have
about DNADynamo functionality. We will get back to you as soon as we can.
-----------------------------------
Copyright © BlueTractorSoftware Ltd, all rights reserved
A) Guide/Reference Mode - to confirm your sequence against the expected
result or find mutations.
For example, you have just PCR sub-cloned Your Favorite Gene (YFG) into a vector, and you need to confirm that no errors have been introduced during the PCR reaction, so you have sent of a few minipreps to your sequencing service together with sequencing primers to sequence in from both ends, and received back a bunch of .abi files. 1) open a DNA Dynamo file that contains the wildtype sequence of YFG in the main sequence window. From the 'Sequencing' menu, select 'Open And Align Sequence Data Files to this Windows Sequence', and then select the .abi files that represent the sequence of one clone. DNA Dynamo will align the sequences in the .abi files to the sequence of the 'guide' dna in the main sequence window, and display the aligned sequences in a sequencing editor window. The result of such an alignment, where two .abi files were aligned against expected sequence, is shown below. Note that DNA Dynamo automatically determines whether the sequence in the abi files is better aligned as forward or reverse - and this is indicated on the graphics map by arrows - though these are not visible at this resolution.







"DNADynamo is by far and away the best and quickest
software for checking sequences"- Professor Greg Towers,
University College London
A guide to basic DNA Dynamo
sequence analysis functions.
So you've sent off some DNA to be sequenced and received back some .abi or .scf files containing the trace data and base calls. Now you want to analyse your sequencing data. DNA Dynamo can help you accomplish this quickly and efficiently, without the need to spend hours analysing each separate data trace manually. By auto assembling translated multiple alignments of your sequencing data, where each base in the alignment is linked to it's chromatogram peak, and presenting an interactive graphic map of sequencing discrepancies, open reading frames and features maps, as well as interfacing with NCBIs Blast and VecScreen services, you can get a handle on your sequence data within minutes. Intuitive sequence editing buttons allow you to manually edit your chromatogram trace base calls and then save the edited and assembled sequence for future reference. By spending a few minutes working out how to use DNA Dynamo for sequence analysis you will save yourself a vast amount of time and effort in the future. DNA Dynamo works in two modes for sequence assembly, depending on whether you know what the expected sequence should be (eg sequencing a construct after subcloning or confirming the result of a site directed mutagenesis experiment) or the sequence is unknown (eg sequencing inserts from a two-hybrid screen, degenerate PCR or other similar library screens).




This video will introduce you to some of the basic sequencing
functions in DNADynamo. You can also read a text based
introduction below, which covers some additional options.
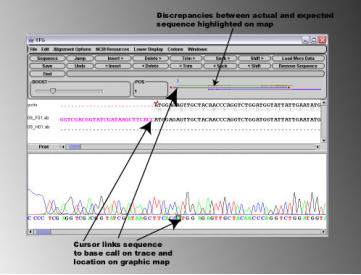
Discrepancies between the expected sequence and the abi
sequence files are highlighted on the graphic map. In the
example shown above, a patch of bad sequence is indicated
with an arrow. Clicking on this section of the graphic map
displays the corresponding sequence and chromatogram
trace in the display areas. It can be seen in the illustration
below that the area of 'bad sequence' is due to the fall off of
sequencing quality at the end of the read, as opposed to 'real'
errors in the sequence. Such ends of bad sequence can
easily be removed using the 'Trim' buttons on the sequence
editing panel.
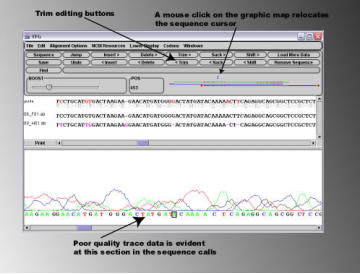
By placing the cursor on the right hand side of the area of low
sequence quality, and pressing the '<Trim' button, the
sequence to the left of the cursor is removed from the
alignment. Note that the characters '<' and '>' present on the
editing buttons indicate on which side of the cursor the editing
effect will take place.
After removing both sections of bad sequence, it can bee
seen that the sequencing data in the .abi files corresponds
exactly to the expected 'guide' sequence.
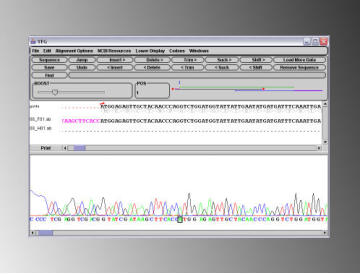
Another useful feature of the sequence editor is the ability to
highlight oligo hits or annotations on the 'guide' sequence.
This can be very useful for example when determining if a
site directed mutagenesis reaction has been successful, as
you can compare the sequence in your .abi file directly to
both the guide sequence and sequence of the mutagenesis
oligo simultaneously.
B) Consensus Mode - assembling unknown sequences.
To open an abi file without aligning the sequence against a guide, simply start from a DNADynamo window that does not contain any sequence in its main sequence window. You may open a single file simply by using the 'Open' option in the 'File' menu. Alternatively you can open multiple files using the 'Import and Align Multiple Files' option under the 'Sequencing' menu. When multiple files are opened, DNA Dynamo attempts to assemble a continuous stretch of sequence (contig) from the separate sequences. The consensus of the assembled sequences is shown together with each individual sequence and trace. In the example shown below, 5 .abi files were assembled to a consensus, and an Open Reading Frame found and set. Stretches of sequence from the consensus can be sent directly to NCBI for Blast analysis. Note that DNA Dynamo is suitable for small scale sequencing projects and is not intended for the assembly of large stretches of sequence.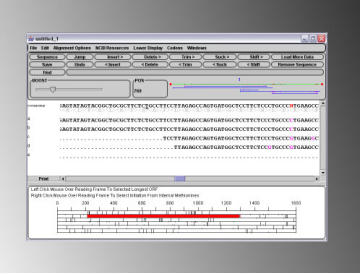
As a point of interest, the first previously unknown sequence
that was assembled and published using DNADynamo's
consensus mode sequence assembly functions was the
African Green Monkey TRIM5alpha gene (Lv1 retroviral
restriction factor activity) in Greg Towers Lab - PNAS
-----------------------------------
DNA Dynamo contains many more functions. See the help
pages for more advice, or feel free to use the 'Ask a
Question' option in the File menu, or email
support@bluetractorsoftware.co.uk directly to ask any
questions you may have about DNADynamo functionality. We
will get back to you as soon as we can.
-----------------------------------
Copyright © BlueTractorSoftware Ltd, all rights reserved
A) Guide/Reference Mode - to confirm your sequence against the
expected result or find mutations.
For example, you have just PCR sub-cloned Your Favorite Gene (YFG) into a vector, and you need to confirm that no errors have been introduced during the PCR reaction, so you have sent of a few minipreps to your sequencing service together with sequencing primers to sequence in from both ends, and received back a bunch of .abi files. 1) open a DNA Dynamo file that contains the wildtype sequence of YFG in the main sequence window. From the 'Sequencing' menu, select 'Open And Align Sequence Data Files to this Windows Sequence', and then select the .abi files that represent the sequence of one clone. DNA Dynamo will align the sequences in the .abi files to the sequence of the 'guide' dna in the main sequence window, and display the aligned sequences in a sequencing editor window. The result of such an alignment, where two .abi files were aligned against expected sequence, is shown below. Note that DNA Dynamo automatically determines whether the sequence in the abi files is better aligned as forward or reverse - and this is indicated on the graphics map by arrows - though these are not visible at this resolution.
DNADynamo
BlueTractorSoftware
For Windows, OSX and Linux